Biology of Neuropathic Pain: A Brief Review
Author
Tony L. Yaksh, PhD
Department of Anesthesiology
University of California, San Diego
Introduction
Following soft tissue injury and inflammation, pain is a common symptom, the disappearance of which is considered to be a consequence of the healing process. In contrast, over time after a variety of injuries to the peripheral nerve, the animal and human will often reflect the appearance of a constellation of pain events. Frequent components of this evolving syndrome are i) ongoing incidences of sharp-shooting sensations referred to the peripheral distribution of the injured nerve and ii) abnormal painful sensations in response to light tactile stimulation of the peripheral body surface. This latter phenomenon is referred to as tactile allodynia. The psychophysics of this state clearly emphasize that the pain is evoked by the activation of low threshold mechano-receptors (Aß afferents). This ability of light touch evoking this anomalous pain state is de facto evidence that the peripheral nerve injury has led to a reorganization of central processing, i.e. it is not a simple case of a peripheral sensitization of otherwise high threshold afferents.
In addition to these behavioral changes, the neuropathic pain condition may display other contrasting anomalies, including on occasion, an ameliorating effect of sympathectomy of the afflicted limb and an attenuated responsiveness to analgesics such as opiates. What are the mechanisms which underlie this nerve injury pain phenotype? This review addresses the principal changes that occur in connectivity and processing after peripheral nerve injury.
Morphological and Functional Correlates of Nerve Injury
The mechanisms underlying this spontaneous pain and the miscoding of low threshold afferent input are not completely understood. As an overview, these events are believed to reflect i) an increase in spontaneous activity in axons in the injured afferent nerve and/or the dorsal horn neurons; and ii) an exaggerated response of dorsal horn neurons to normally innocuous afferent input.
Following peripheral nerve ligation or section, several events occur signaling long-term changes in peripheral and central processing. Thus, in the periphery after an acute mechanical injury of the peripheral afferent axon, there will be an initial dying back (retrograde chromatolysis) that proceeds for some interval at which time the axon begins to sprout sending growth cones forward. The growth cone frequently fails to make contact with the original target and displays significant proliferation. Collections of these proliferated growth cones form structures called neuromas. Examination of the cell body of these injured axons reveals that though they are not injured, they display evident cellular responses indicative of injury, including increased protein expression.
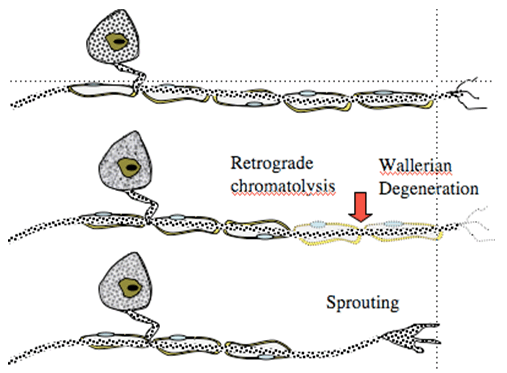
Figure 1. Nerve injury leads to degeneration of distal terminal and dying back (retrograde chromatolysis). Overtime, sprouting occurs.
1. Spontaneous Pain State
Under normal conditions, primary afferents show little if any spontaneous activity. Following an acute injury to the nerve, afferent axons will display i) an initial burst of afferent firing secondary to the injury; ii) silence for an interval of hours to days; and iii) followed over time by the development of a measurable level of spontaneous afferent traffic in both myelinated and unmyelinated axons. This ongoing input is believed to provide the source of the afferent activity that leads to spontaneous on going sensation. (Figure 2).
a. Site of Origin of Spontaneous Afferent Traffic
Single unit recording from the afferent axon has indicated that the origin of the spontaneous activity in the afferent arises from the neuroma and from the dorsal root ganglia of the injured axon. Activity in sensory afferents originates after an interval of days to weeks from the lesioned site (neuroma) and from the dorsal root ganglion (DRG) of the injured nerve. (Figure 2).
b. Mechanisms of Ectopic Activity
Following peripheral nerve injury there is a very significant upregulation in the expression of a wide variety of proteins in the dorsal root ganglion and thus the axon and terminals of the injured primary afferent. Among these protein which show upregulation are a variety of channels and receptors.
Increased Channel Expression
Two specific examples will be noted:
- Voltage sensitive sodium channels mediate the conducted potential in myelinated and unmyelinated axons. Cloning has emphasized that there are multiple populations of voltage sensitive sodium channels (NaV), differing in their current activation properties and structure. This increased ionic conductance may result in the increase in spontaneous activity that develops in a sprouting axon. Concentrations of sodium channel blockers such as lidocaine which will not block conduction will attenuate ectopic activity in the dorsal root ganglia and neuroma. At these concentrations IV lidocaine will diminish the hyperpathic behavior in animal and human models of nerve injury.
- The alpha2delta subunit of the Voltage gated calcium channel also plays an important role. After nerve injury increased expression of this subunit has been identified in the DRG. This subunit has been shown to be the binding site for agents such as gabapentin. Gabapentin has been shown to attenuate hyperpathic behavior in human and animal models of nerve injury (Figure 3).
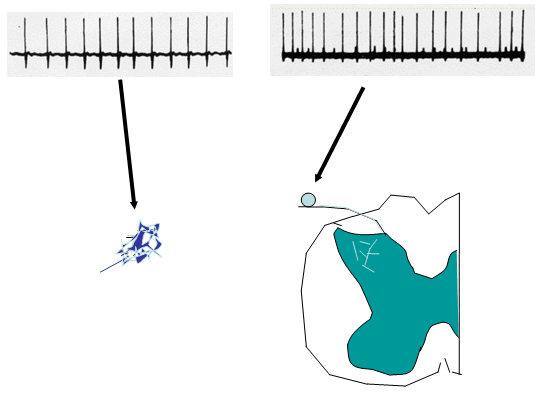
Figure 2. Following peripheral nerve injury, injured axon sprout to form a neuroma. These neuromas become ectopic generators of neural activity. In addition, the dorsal root ganglion cells of these injured axons also demonstrate ongoing discharges in the injury environment.
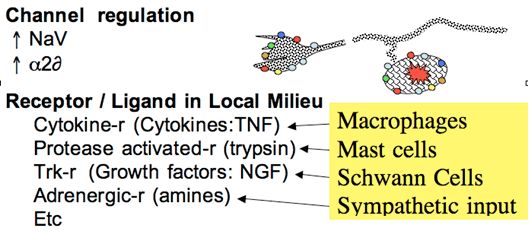
Figure 3. After injury, ectopic activity is believed to arise from increased expression of a variety of channels and receptor protein. These receptors are sensitive to the products that are found in the injury environment of the neuroma and the DRG to yield local depolarization.
Changes in Afferent Terminal Sensitivity
The sprouted terminals of the injured afferent axon display a characteristic growth cone that possesses transduction properties that were not possessed by the original axon. These include significant mechanical and chemical sensitivity. Thus, these sprouted endings, as well as the DRG of the injured axons may display increased expression of a variety of receptor proteins such as TRK receptors (for growth factors such as NGF), prostanoid receptors for a variety of lipid mediators, adrenergic receptors for catecholamines, and cytokines receptors for agents such as TNF. This evolving sensitivity is of particular importance given that following local nerve injury there is the release of these products from Schwann, macrophages and mast cells. Such agents, particularly TNF can thus directly activate the DRG and neuroma. In addition, following nerve injury, there is an important sprouting of postganglionic sympathetic efferents that can lead to the local release of catecholamines. This scenario is consistent with the observation that following nerve injury, the postganglionic axons can initiate excitation in the injured axon (see below). These events are believed to contribute to the development of spontaneous afferent traffic after peripheral nerve injury (Figure 3).
2. Evoked Hyperpathia
The observation that low threshold tactile stimulation yields a pain state has been the subject of considerable interest. As noted, there is considerable agreement that these effects are often mediated by low threshold (Aß) afferent stimulation. Several underlying mechanisms have been proposed to account for this seemingly anomalous linkage.
a. Dorsal Root Ganglion Cell Cross Talk
Following nerve injury, evidence suggests that “cross-talk” develops between afferents in the DRG and in the neuroma (Figure 4). Here, depolarizing currents in one axon would generate a depolarizing voltage in an adjacent quiescent axon. This depolarization would permit activity arising in one axon to drive activity in a second. In this manner, it is hypothesized that a large low threshold afferent would drive activity in an adjacent high threshold afferent. Alternatively, dorsal root ganglion cells in vitro can release a variety of transmitters and express excitatory receptors.
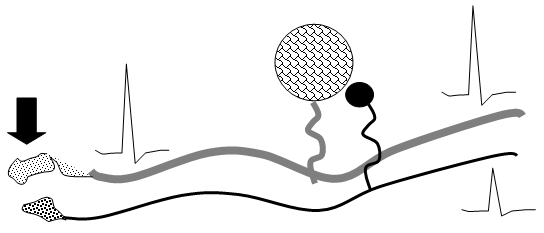
Figure 4. Schematic indicating the functional cross talk which may occur between primary afferent axons (Aß and C fibers) which have been injured, at the site of injury (neuroma) and at the level of the DRG.
b. Afferent Sprouting
Under normal circumstances, large myelinated (Aß) afferents project into the spinal Rexed Lamina III and deeper. Small afferents (C-fibers) tend to project into spinal laminae II and I (a region consisting mostly of nocisponsive neurons). Following peripheral nerve injury, it has been argued that the central terminals of these myelinated afferents (A-fibers) sprout into lamina II of the spinal cord. With this synaptic reorganization, stimulation of low threshold mechano-receptors (Aß fibers) could produce excitation of these neurons and be perceived as painful. The degree to which this sprouting occurs is a point of current discussion and while it appears to occur, it is much less prominent than originally reported.
c. Dorsal Horn Reorganization
Following peripheral nerve injury, a variety of events occur in the dorsal horn, which suggests altered processing wherein the response to low threshold afferent traffic can be exaggerated. It should be stressed that the ability of Aß afferents to evoke pain behavior after nerve injury reflects several conundrums. First, Aß axons are known to innervate dorsal horn wide dynamic range (Lamina V) neurons. These neurons are known to also receive input from A∂ and C fibers and are believed to play a role in the pain encoding. Yet, Aß input normally does not cause pain. There may be several reasons for this, but increasingly there is an emphasis being placed upon dorsal horn reorganization and the role played by interneuronal systems that clearly regulate large afferent excitability.
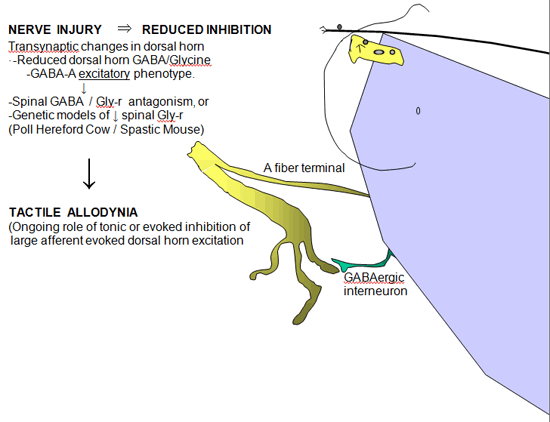
Figure 5. Schematic indicating the effects of reduced GABA A ergic and glycinergic inhibition in the dorsal horn after nerve injury consequence of increased glutamate release in the dorsal horn secondary to nerve injury.
Loss of Intrinsic GABAergic/Glycinergic Inhibitory Control
In the spinal dorsal horn there are a large number of small inter-neurons that contain and release GABA and glycine. GABA / glycinergic terminals are frequently presynaptic to the large central afferent terminal complexes and form reciprocal synapses, while GABAergic axosomatic connections on spinothalamic cells have also been identified (Figure 5). These amino acids normally exert an important tonic or evoked inhibitory control over the activity of Aß primary afferent terminals and second order neurons in the spinal dorsal horn. The relevance of this intrinsic inhibition to pain processing is provided by the observation that the simple intrathecal delivery of GABA A receptor or glycine receptor antagonists will lead to a powerful behaviorally defined tactile allodynia. Similarly, animals genetically lacking glycine-binding sites often display a high level of spinal hyper-excitability. These observations led to consideration that following nerve injury there may be a loss of GABAergic neurons.
Change from Inhibitory to Excitatory Actions of GABA Synapses
While there are data that do support a loss of spinal GABAergic neurons, the loss appears to be minimal at best. Recent observations now suggest a second alternative. After nerve injury, spinal neurons regress to a neonatal phenotype in which GABA-A activation becomes excitatory. A recent insight is that afferent nerve injury leads to a reduction in the expression of Cl transporter protein in dorsal horn neurons. Under normal conditions transmembrane [Cl-] are at equilibrium at or just below resting membrane potentials. Increasing Cl permeability by GABA-A or glycine receptor (Cl- channels) yields hyperpolarization and inhibition. “Cation-Cl” cotransporters regulate Cl- gradient by exporting [Cl-]i The loss of dorsal horn DHN-KCC2 after nerve injury leads to increased intracellular [Cl-]i Under these conditions, increasing Cl permeability may lead to a failure of GABA-A / glycine inhibition or in fact turning the GABA/glycine effect into excitation of the 2° neuron (Figure 6).
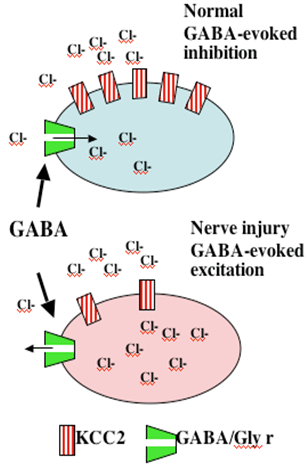
Figure 6. Following peripheral nerve injury, there is a loss of Cl transporter, leading to accumulation of intracellular Cl. Now, increasing Cl permeability as by opening the GABA-A or glycine ionophore may have no inhibitory effect or may lead to an excitatory potential.
Spinal Glutamate Release
There is little doubt that the post-nerve injury pain state is dependent upon an important role of spinal glutamate release (Figure 7). Recent studies have emphasized that after nerve injury there is a significant enhancement in resting spinal glutamate secretion. This release is in accord with i) an increased spontaneous activity in the primary afferent, and ii) with the loss of intrinsic inhibition that may serve to modulate resting glutamate secretion (see below). The physiological significance of this release is emphasized by several convergent observations.
- Intrathecally delivered glutamate will evoke a powerful tactile allodynia and thermal hyperalgesia though the activation of spinal NMDA and non-NMDA receptors.
- The spinal delivery of NMDA antagonists has been shown to attenuate the hyperpathic states arising in animal models of nerve injury. NMDA receptor activation mediates an important facilitation in neuronal excitability. In addition, the NMDA receptor is a Calcium ionophore which when activated leads to prominent increases in intracellular calcium. This increased calcium serves to initiate a cascade of events that includes the activation of a variety of enzymes (kinases) some of which phosphorylate membrane proteins (e.g. calcium channels and the NMDA receptors) while others such as the mitogen activated kinases (MAP kinases) serve to mediate intracellular signaling that leads to the altered expression of a variety of proteins and peptides (e.g. cyclooxygenase and dynorphin). This downstream nuclear action is believed to herald long term and persistent changes in function. A variety of factors have been shown to enhance glutamate release.
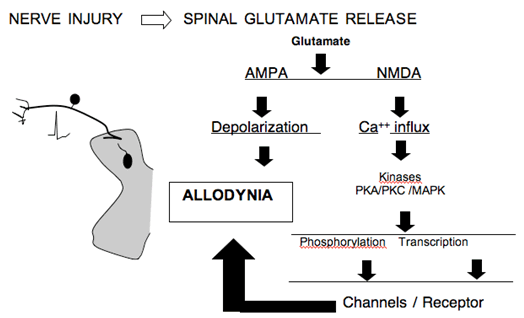
Figure 7. Schematic indicating the consequence of increased glutamate release in the dorsal horn secondary to nerve injury.
Non-neuronal Cells and Nerve Injury
Following nerve injury (section or compression), it has been shown that there is a significant time dependent increase in activation of spinal microglia and astrocytes in the spinal segments receiving input from the injured nerves. Of particular interest is that in the face of pathology such as bone or pancreatic cancer, such upregulation has been clearly shown. Astrocytes are activated by a variety of neurotransmitters and growth factors. While the origin of this activation is not clear, it will lead to an increased spinal expression of COX /NOS / Glutamate transporters / Proteinases. Such biochemical components have been previously shown to play an important role in the facilitated state.
Sympathetic Dependency of Nerve Injury Pain State
After peripheral nerve injury, there is an increased innervation of the peripheral neuroma by post-ganglionic sympathetic terminals. More recently, it has been show that there is an in-growth of post-ganglionic sympathetic terminals into the dorsal root ganglia of the injured axons. These post-ganglionic fibers form baskets of terminals around the ganglion cells. Several properties of this innervation are interesting (Figure 8).
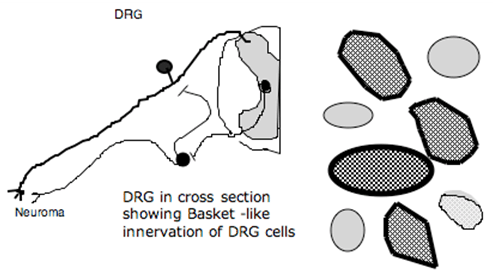
Figure 8. Schematic showing sprouting into neuroma and into the DRG after nerve injury. Post-ganglionic axons form baskets around DRG cells. Activation of ventral roots (containing preganglionic sympathetic axons) results in activation of activity from neuroma and DG.
- They invest all size ganglion cells, but particularly Type A (large ganglion cells);
- The innervation occurs principally in the DRG ipsilateral to the lesion, but in addition, there is innervation of the contralateral ganglion cell. Stimulation of the ventral roots of the segments, containing the pre-ganglionic efferents, will produce activity in the sensory axon by an interaction either at the peripheral terminal at the site of injury or by an interaction at the level of the DRG. This excitation is blocked by intravenous phentolamine, emphasizing an adrenergic effect. Whether these effects are indeed underlying sympathetic pain states is an issue of controversy.
Summary
- Classic observations emphasize that nerve section leads to painful sensory experiences that reflect the persistence of a neural organization that originally subserved that body region innervated by the sectioned nerve.
- Pain is associated with ongoing sensations (dysesthesias) and an exaggerated sensitivity to otherwise innocuous stimuli (e.g. tactile allodynia).
- Allodynia represents activity in low threshold afferents. At a systems level, nerve injury results in increases in afferent traffic from the injured nerve at the site of injury (e.g. the neuroma) and/or at the dorsal root ganglion of the injured axons.
- Injury also produces alterations in the dorsal horn excitability leading to enhanced activity in dorsal horn neurons and changes in their receptive field size.
- A number of mechanisms have been identified which are believed to underlie these changes in dorsal horn excitability, including increased expression of pro excitatory element such as sodium and calcium channels and the increased expression of cytokines and their eponymous receptors.
- In addition, there are long term changes in receptor properties such as the conversion of the GABA A receptor from an inhibitory to an excitatory phenotype secondary to changes in the Chloride transporter.
- Activation of nonneuronal cells are also considered to be relevant where in they serve to release a variety of proexcitatory products.
- Sprouting of primary afferents does not explain the pain events secondary to injury, but the sprouting of sympathetics into the DRG and neuroma may provide a linkage for the sympathetic component of the sympathetic component of the pain state.
References
- Calcutt NA, Backonja MM.Pathogenesis of pain in peripheral diabetic neuropathy. Curr Diab Rep 2007;7:429-34
- Day M. Sympathetic blocks: the evidence. Pain Pract 2008;8:98-109.
- Hansson PT, Dickenson AH. Pharmacological treatment of peripheral neuropathic pain conditions based on shared commonalities despite multiple etiologies. Pain 2005;113:251-4.
- Romero-Sandoval EA, Horvath RJ, DeLeo JA. Neuroimmune interactions and pain: focus on glial-modulating targets. Curr Opin Investig Drugs 2008;9:726-34.
- Saadé NE, Jabbur SJ. Nociceptive behavior in animal models for peripheral neuropathy: Spinal and supraspinal mechanisms. Prog Neurobiol 2008;86:22-47.
- Whiteside GT, Adedoyin A, Leventhal L. Predictive validity of animal pain models? A comparison of the pharmacokinetic-pharmacodynamic relationship for pain drugs in rats and humans. Neuropharmacology 2008;54:767-75.
- Woolf CJ. Dissecting out mechanisms responsible for peripheral neuropathic pain: implications for diagnosis and therapy.Life Sci 2004;74:2605-10.
- Yaksh TL, Sorkin LS. Mechanisms of neuropathic pain. Curr Med Chem 2005;5:129-140.
Leave a commentOrder by
Newest on top Oldest on top