Central Post-Stroke Pain
Authors
Shao-Lun Tsao, MD
Staff Anesthesiologist
Department of Anesthesiology
Division of Pain Management
Changhua Christian Hospital
Changhua, Tawian
Young Woo Cho, MD, PhD
Associate Professor
Department of Anesthesiology and Pain Medicine
Ulsan University Hospital
Ulsan, Korea
Billy Huh, MD, PhD
Professor and Medical Director
The University of Texas M.D. Anderson Cancer Center
Department of Pain Medicine
Houston, TX
Introduction
Post-stroke pain (PSP) was first described by Dejerine and Roussy in 1906 as “thalamic syndrome,” but it is well known that PSP is not limited to lesions in the thalamus. PSP can be triggered by injury to other areas of the central nervous system.[1] Moreover, incidence of chronic pain following stroke varies widely among literature. The fact that pain is not always associated with stroke, the presence of different types of PSP and the ubiquity of pre-existing chronic pain in this population make diagnosis of PSP challenging. The most common forms of PSP are musculoskeletal pain, central post-stroke pain (CPSP), headache, as well as pain from spasticity.[2-3]
CPSP is a central neuropathic pain syndrome that occurs after an ischemic or hemorrhagic stroke and can affect tissue anywhere along the spinothalamic pathway, thalamus, or its cortical projections. It is often overlooked due to the presence of other more prominent neurologic symptoms following a stroke e.g., sensory deficit, inability to move a limb, or, inability to compose or understand speech. CPSP is not a rare pain syndrome affecting 5-8% of patients after a stroke, but the prevalence varies depending on the location of the lesion. Clinical data have demonstrated that patients with lateral medullary infarction (Wallenberg’s syndrome), or thalamic infarction involving the ventral caudal nucleus are more prone to develop CPSP.[4-5]
Clinical Presentation
There are no particular pathognomonic features of CPSP, and clinical presentations are similar to other central or peripheral neuropathic pain syndromes. Due to the lesion in the sensory pathway, sensory impairment to touch, pinprick and temperature is typically observed immediately after stroke. Onset of pain gradually occurs within a smaller area than the region of sensory impairment, especially within the region of cold sensory deficit. Spontaneous pain is continuous and/or paroxysmal, with most commonly described qualities of burning, lacerating, aching and pricking. Hyperalgesia to noxious stimuli (thermal, punctuate), as well as allodynia to innocuous stimuli (cold, light touch, movement), are commonly manifested in patients with CPSP (Table 1). A majority of patients also have autonomic instability, including hyperhidrosis, cutaneous blood flow change, and pain exacerbation by physical and emotional stress.[6-8]
Table 1. Symptoms of CPSP[6-8]
| Symptoms | Descriptions |
|---|---|
| *Allodynia affects two-thirds of CPSP patients | |
| Spontaneous pain (continuous and/or paroxysmal) | Burning, lacerating, aching, pricking, squeezing, shooting, throbbing, heaviness, freezing |
| Allodynia* | Innocuous cold, light touch, movement |
| Hyperalgesia | Noxious thermal, punctate |
| Sensory abnormality | Temperature, touch, pinprick |
| Autonomic instability | Regional blood flow change, hyperhidriosis, pain aggravated by emotional and physical stress, and relieved by rest and relaxation |
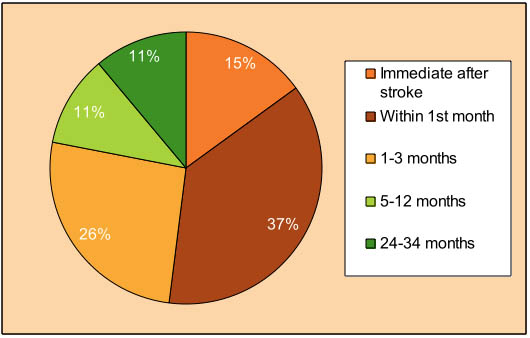
Figure 1. Time interval between stroke and CPSP
The distribution of pain and sensory deficit can develop in the limbs, trunk, and face on the side contralateral to the cerebral lesion, except for patients with lateral medullary infarctions where facial pain and sensory deficit are occasionally ipsilateral.4 The area of the distribution varies among individuals, and can range from a small area (e.g., a leg, or a hand) to large areas (e.g., hemi-body pain).8 Some patients may present delayed-onset sensory symptoms that can be mild and without objective sensory deficit, developing at the body parts mirroring the site of the most severe central post-stroke pain.[9]
The onset of CPSP is usually delayed and gradual after the stroke episode. Only approximately 15% of CPSP patients have pain symptoms immediately after stroke, and 75% of the patients develop CPSP within 6 months (Figure 1).[2][6] The mechanism of delayed pain onset may be attributed to slow biochemical or anatomical changes following the stroke,[10] i.e. axonal sprouting, regrowth and collateral sprouting;[11] effects induced by apoptosis and other forms of slowly developing cell death;[12] gradual development of collateral pathways and unmasking of pre-existing connections;[13] and slowly evolving depletion or activation of receptors or ion channels.[14]
Diagnosis
There are no universally accepted criteria for the diagnosis of CPSP. The diagnosis is based on the combination of patient history, clinical and sensory examination, exclusion of other causes of pain, and confirmatory CT or MRI of the relevant lesion. The common causes of chronic post-stroke pain should be excluded in any analysis of CPSP include musculoskeletal pain, shoulder pain, tension-type headache, and painful spasticity.[3][15] Unfortunately, chronic pain is often a preexisting symptom in this patient population, thereby further complicating the clinical diagnosis (Figure 2).
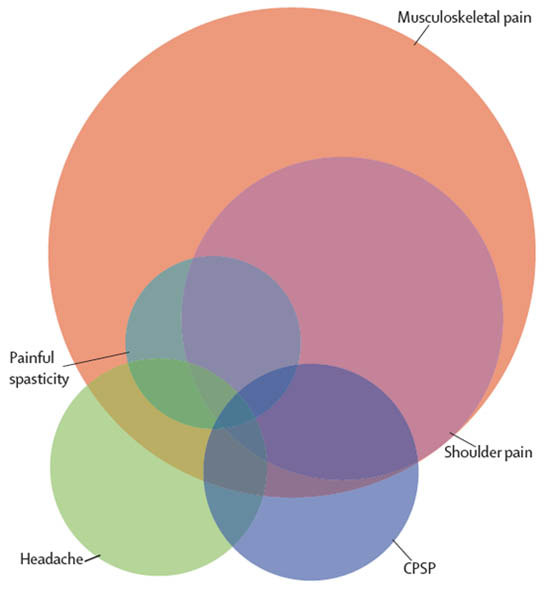
Figure 2. Common types of chronic pain that can occur after stroke[18] Reprint permission granted from publisher
Individual patients can have various combinations of one or several types. The size of the circles is approximate to relative frequency (spasticity 7%, headache 10%, CPSP 10%, shoulder pain 20%, and musculoskeletal 40%).
Other laboratory measures, such as somatosensory-evoked potentials (SEPs) and laser-evoked potentials (LEPs), are generally found to be abnormal in patients with CPSP,[16-17] but their clinical use is debatable. Klit et al. have proposed a grading system (possible, probable, definite) for diagnosis of CPSP based on 5 criteria shown in Table 2, but this method does not exclude other causes of pain.[18]
Table 2. Grading system for CPSP Pathophysiology by Klit et al[18]
| CPSP is defined as "possible" if criteria 1, 2, and 3 are fulfilled, "probable" if criteria 1, 2, and 3 plus either criteria 4 or 5 are fulfilled, and "definite" if criteria 1–5 are fulfilled. | |
|---|---|
| 1 | Exclusion of other likely causes of pain No other obvious cause of pain |
| 2 | Pain with a distinct neuroanatomically plausible distribution Either pain is localized unilaterally in the body an/or face or unilaterally on one side of the body with contralateral involvement of the face |
| 3 | A history suggestive of stroke Sudden onset of neurological symptoms with onset of pain at or after stroke onset |
| 4 | Indication of the distinct neuroanatomically plausible distribution by clinical neurological examination Findings of positive or negative sensory signs in the painful area on clinical examination, pain localized within a territory of sensory abnormality, and anatomically plausible distribution of sensory abnormalities |
| 5 | Indication of the relevant vascular lesion by imaging Visualization of a lesion that can explain the distribution of sensory findings (either CT or MRI) |
Pathophysiology
The mechanism of CPSP is complex and still largely unexplained. Injury to any part of the somatosensory pathway, thalamus, or cortex can result in profound changes in circuitry, and may lead to development of CPSP. Unlike peripheral deafferentation pain, there is no animal model for post-stroke central neuropathic pain. Using the syringomyelia model, Ducreux et al.[19] demonstrated that different central neuropathic symptoms can be attributed to distinct mechanisms. The most plausible mechanism for the central neuropathic pain appears to be the thermosensory disinhibition between the lateral and medial thalamus. The lateral system consists of A-delta fibers from the neospinothalamic tract to the sensory nucleus of the lateral thalamus, with projection to the primary somatosensory cortex, secondary somatosensory cortex and insula. The medial system is composed of C-fibers from the paleospinoreticulothalamic tract to the medial and intralaminar nuclei of the medial thalamus, and projection to the anterior cingulate cortex.
A post-stroke lesion in the neospinothalamic tract can result in partial deafferentation of the lateral system, leading to sensory deficits. Based on an animal model, Craig et al.[20] suggested that central pain following spinothalamocortical pathway injury is attributed to a loss of normal pain inhibition by cold. The loss of cold stimuli input to the insula cortex can disrupt thermoregulatory homeostasis. Subsequently, the diminished thermoregulatory input to the periaqueductal gray and parabrachial nuclei causes disinhibition of the medial pain system, resulting in hyperactivity of the anterior cingulate cortex, leading to development of CPSP.
Other studies have shown that thalamic hyperexcitability is also implicated in the onset of CPSP. Thalamus hyperexcitability after deafferentation is associated with dysregulation of sodium (NaV1.3) and calcium channels (CaV3.1), activation of NMDA receptors, and loss of GABA-ergic neuron innervation. Thus, a sensitized thalamus can function as a pain amplifier, or even as a pain generator.[21-23]
Based on quantitative sensory tests of patients with central neuropathic pain below the spinal lesion, Wasner et al.[24] suggested inflammation and degeneration of injured axons may trigger spontaneous activity in remaining intact neurons as a cause of central pain. However, spinothalamic tract (SST) lesions may be necessary, but not sufficient for development of central pain, since equally severe SST deficit was observed in patients with or without pain.[25] Using isotope[C11]-tagged diprenorphine binding and PET scan, Jones et al.[26] showed significantly reduced opioid receptor binding in the medial pain system (dorsolateral and anterior cingulate, insula cortex, and thalamus) in patients with post-stroke central neuropathic pain. This may explain the reduced opioid response in these patients.
Treatment
The management of CPSP remains a challenge as treatment options are limited with no uniformly effective therapies. In addition to managing pain, improving quality of life and reducing psychological co-morbidity are an integral part of long-term management goals. Here pharmacological and interventional approaches are discussed.
Pharmacological Therapy
Antidepressants
Tricyclic antidepressants (TCAs) have a well-established beneficial effect in various neuropathic pain states, and are considered the first-line drug therapy.[27] TCAs have specific analgesic activity independent of their antidepressant effect.
Amitriptyline was the first drug proven to be effective in a double-blind, placebo-controlled crossover study.[28] In 15 patients with post-stroke pain, amitriptyline was superior to carbamazepine (CBZ) and placebo. Higher plasma concentration of amitriptyline was associated with greater relief from CPSP. The initial treatment is recommended to start at a low dose, 10-20 mg/day, titrated upward weekly to a dose that results in relief or intolerable side effects. The onset of efficacy is approximately 4-7 days after reaching the optimal dose. TCA is limited by adverse effects such as dry mouth, drowsiness and constipation, as well as more rare instances of urinary retention, orthostatic hypotension and cardiac arrhythmia.[29] The role of amitriptyline in the prophylaxis of CPSP has also been evaluated in another study, but the results demonstrated no beneficial effects.[30]
The selective serotonin reuptake inhibitor fluvoxamine up to 125 mg showed some efficacy in an open-labeled study of 31 patients with CPSP. The average pain level was reduced from 7.7 to 6.0, determined on a visual analog scale. It was effective in those patients who had stroke within 1 year, and was not related to its antidepressant action.[31]
Anticonvulsants
Anticonvulsants are the second most widely prescribed drugs in the management of central pain. These drugs reduce abnormal neuronal hyperexcitability through modulation of sodium or calcium channels, effect on excitatory amino acids, and GABA-mediated disinhibition. Although CBZ was less effective than amitriptyline, CBZ has been shown to be effective in paroxysmal shooting pain related to the CNS. However, the use of CBZ has been limited by its side effect profile, and by the need for very gradual dose escalation, especially in the elderly. Dizziness and somnolence are the most commonly reported adverse effects.[28][32]
A randomized control trial of lamotrigine showed significant analgesia in patients with CPSP with a mean reduction of spontaneous pain by 30%. A dose of lamotrigine 200 mg/day reduced the median pain score to 5, compared to 7 with placebo in the intention-to-treat population of 27 patients.[33] Also presented in this study, lamotrigine was effective for cold-induced allodynia, but not for mechanical-induced allodynia. Side effects were relatively mild except for rash and headache. However, the potential for more severe side effects such as Stevens-Johnsons and Lyells Syndrome may limit its broad use in neuropathic pain.
Gabapentin has been tested in a randomized, placebo-controlled trial of 305 patients with chronic pain, where 9 presented CPSP. The results showed improvement of pain score in 21% of patients compared to 14% in the placebo group. However, the efficacy was not statistically significant.[34] A pilot study by Attal et al.[35] showed that gabapentin was effective in several pain components including pain paroxysms, and brush/cold-induced allodynia related to both peripheral and central lesions. The most common side effects of gabapentin were dizziness and somnolence which were well tolerated and transient.
Pregabalin has also been shown to have clinically significant analgesic effects on patients with central neuropathic pain in a randomized placebo controlled study by Vranken.[36] Pregabalin not only reduced pain intensity, but also improved quality of life. The most commonly experienced side effects were dizziness, decreased intellectual performance, somnolence, and nausea.
Other anticonvulsants including phenytoin, zonisamide, and topiramate have been tested in studies using a small number of patients with CPSP. Phenytoin was tried in 8 patients with thalamic pain. The dose of phenytoin increased until the side effects appeared. There was marked improvement in 3 patients, 2 minimally, and 3 patients worsened.[37] Zonisamide was found to be useful for 2 patients with posterolateral thalamic infarcts.[38] Topiramate has not been found to be useful in central or neuropathic pain.[39]
Opioids
In a double-blind, placebo-controlled crossover study by Attal et al.[40] 15 patients with central pain (6 with CPSP, 9 with spinal cord injury) were given intravenous (IV) morphine (mean dose 16 mg; range 9-30 mg) and subsequently maintained on oral morphine. Morphine significantly reduced the intensity of brush-induced allodynia but had no effect on other evoked pains, such as static mechanical and thermal allodynia/hyperalgesia. The effects of morphine on ongoing pain were not significantly different from those of the placebo. Although efficacy of IV morphine correlated well with oral morphine up to 1 month, only 3 individuals (20%) were still using oral morphine after 1 year. Most of the patients were withdrawn from the study because of significant side effects from oral morphine, i.e. somnolence, nausea, headache. Hence, morphine showed analgesic effects on some components of central neuropathic pain syndromes, but a limited number of patients may benefit from long-term opioid treatment.
Levorphanol is an opioid with a weak NMDA-receptor antagonist activity. In one study, 81 patients (10 with CPSP) were randomly assigned to high-dose (15.75 mg/day) and low-dose (3.15 mg/day) treatment groups for an 8-week period. Only 3 of 10 patients with CPSP could complete the study because of side effects, and no beneficial effect was observed in these patients.[41] Naloxone was demonstrated to be of no value in alleviating the pain of CPSP in a double-blind, placebo-controlled crossover study.[42] In a case report by Iranami et al.,[43] an IV tramadol infusion was reported to be effective in a patient with CPSP of 10 years duration, refractory to CBZ and amitriptyline. After IV injection of tramadol, complete pain relief was achieved for 5 hours. The patient was asymptomatic for 10 months with codeine phosphate and milnacepram. There were no reported side effects.
Methadone, which has weak NMDA-receptor antagonist and tricyclic antidepressant activity in addition to the µ-agonist effect, may have some role in patients with CPSP, but currently there is no evidence to support wide use.
N-Methyl-d-Aspartate (NMDA) Antagonists
An IV ketamine infusion followed by oral ketamine titrated to 50 mg three times a day was beneficial in decreasing allodynia and hyperalgesia, as well as improving functional capabilities.[44] In an uncontrolled trial, 11 of 23 patients with CPSP receiving IV ketamine hydrochloride (5 mg every 5 min, up to a total dose of 25 mg) prior to chronic motor cortex stimulation reported pain reduction of at least 40%, lasting less than 1 hour.[45]
The NMDA antagonist dextromethorphan was effective in diabetic neuropathy but showed no efficacy in central pain.[46]
Antiarrhythmics
Lidocaine, a sodium channel blocker, is the most effective agent available for central pain when administered intravenously. In a randomized, double-blind, placebo-controlled trial on 16 subjects with central pain, 6 of whom had CPSP, lidocaine was notably superior to the placebo in reducing the intensity of spontaneous ongoing pain for up to 45 min after the injection. Of those tested, 10 patients (63%) receiving lidocaine showed a significant reduction in spontaneous pain, whereas only 6 patients (38%) showed improvement with the placebo. Lidocaine also reduced the intensity of brush-induced allodynia and mechanical hyperalgesia, but was no better than the placebo against thermal-induced allodynia and hyperalgesia. The side effects were moderate, consisting mainly of lightheadedness. Three weeks after completion of the study, 12 patients received mexiletine 200 mg/day, then titrated up to 400-800 mg/day. Mexiletine, which is an oral analogue of lidocaine was not as effective as IV lidocaine in the management of central pain. The results indicated that 4 patients experienced slight to moderate pain relief, and 8 patients reported no pain relief.[47]
GABA-ergic Drugs
Propofol is a GABA-a agonist and has been investigated in a double-blind, placebo-controlled study, where 32 patients with central pain (7 with CPSP) were given a single bolus, sub-hypnotic dose (0.2 mg/kg) of IV propofol. If the patients responded to bolus tests (decreased allodynia) , they were given infusion of propofol 0.3 mg/kg/h for 6-24 h. Spontaneous pain and allodynia were relieved in 5 out of 7 patients with CPSP, and the analgesic effect was maintained for 4-12 hours after infusion. However, neither propofol nor placebo reduced central pain. Propofol may therefore help in differential diagnosis, as well as short-term treatment.[48]
The IV GABA-a agonist thiopental when used in subanesthetic doses (maximum 250 mg) showed pain reduction of at least 40% in 22 of 39 patients with CPSP in an uncontrolled study. The pain-reducing effects lasted approximately 1 h.[45]
Thiamylal is another GABA-a agonist, and was investigated in an uncontrolled study. In the study, 39 patients with CPSP were given 50 mg of thiamylal every 5 min up to the total dose of 250 mg. Of the 39 patients, 22 reported pain reduction of more than 40%, and the efficacy lasted more than 1 h.[45]
Baclofen is a GABA-b agonist, and its intrathecal administration has been investigated in an open-label study. A bolus of intrathecal baclofen (50-100 mcg) was given to 14 patients with central pain (8 with CPSP). It was reported that 6 of the 8 patients with CPSP had substantial pain relief 1-2 h after injection, and relief lasted for 10-24 h.[49] Further investigation will be needed to determine if an implantable intrathecal device with baclofen is helpful for patients with CPSP.
Interventional Procedures
Neurostimulation
Non-pharmacological therapies of CPSP have been focused mainly on neuromodulation techniques such as motor cortex stimulation (MCS), deep brain stimulation (DBS), repetitive transcranial magnetic stimulation (rTMS), and vestibular caloric stimulation (VCS). They are usually indicated for treatment-resistant cases of CPSP. There are only a few randomized, placebo-controlled studies of neurostimulation therapy for CPSP and published papers largely consist of case series and case reports.
Motor Cortex Stimulation (MCS)
The MCS technique consists of implanting epidural electrodes over the motor strip through a frontoparietal craniotomy over the central area, under general anesthesia, or, through a simple burr hole under local anesthesia. The mechanism of action of MCS remains hypothetical, although there is supporting evidence from animal and human trials. The fact that many of the regions activated by MCS containing high levels of opioid receptors suggest that long-lasting MCS effects may also involve secretion of endogenous opioids.[50-51]
A long-term follow-up study by Nguyen et al.[52] on 32 MCS patients with post-stroke pain reported 15 patients (48%) with excellent to good pain control (pain reduction > 60%) at intensities below the threshold for muscle contraction. Satisfactory pain control was achieved in 13 (73%) of 18 patients in whom motor weakness in the painful area was virtually absent or mild. Only 2 (15%) of the 13 patients who had demonstrated moderate or severe weakness in the painful area received analgesia. Based on a review of the current literature, Cruccu et al.[53] stated that the overall success rate of MCS was in the range of 50-60% for patients with CPSP. The most common complications of treatment by MCS are seizure, infection, hardware problems, and extradural hematoma.[54]
Deep Brain Stimulation (DBS)
DBS for the treatment of medically refractory chronic pain preceded the gate theory. But the mechanism by which DBS relieves pain remains unclear. The main targets of deep brain stimulation in patients with CPSP are the sensory (ventral posterior) thalamus, and the periventricular gray matter. After accurate target localization using MRI, stereotactic computerized tomography and brain atlas co-registration as appropriate, an electrode is stereotactically inserted into the subcortical cerebrum under local anesthesia. The advantages of DBS are that it is reversible, nondestructive and can be modified by adjustment of the stimulator settings after implantation. DBS in CPSP patients had efficacy rates ranging from 25% to 70%.[55-56]
A meta-analysis by Bittar et al.[57] showed that DBS was more effective for nociceptive pain, rather than for deafferentation pain (63% vs 47%, respectively). The stimulation was successful in approximately 50% of those with post-stroke pain, and 58% of patients permanently implanted achieved ongoing pain relief. However, only 31% of the patients with a central lesion experienced satisfactory long-term pain relief (p<0.03). The higher rates (51%) of success were seen with peripheral neuropathies. An extensive literature review by Cruccu et al.[53] from 1968 to 2006 cited that efficacy of DBS on CSPS is equivocal, and requires further comparison trials.
Repetitive Transcranial Magnetic Stimulation (rTMS)
Repetitive Transcranial Magnetic Stimulation (rTMS) is a noninvasive motor cortex stimulation technique that has been shown to be effective in CPSP, and may produce long-lasting pain relief. The rTMS is performed by applying the coil of a magnetic stimulator on the scalp, above a targeted cortical region. The rationale is same as for an implanted MCS. It can be applied to any patient with refractory neuropathic pain who may be a good candidate for the cortical stimulator.
A randomized, controlled study by Khedr58 showed that rTMS resulted in significant reduction in pain compared to a control group, and the pain relief was sustained for 2 weeks. There were no significant side effects. Lefaucheur et al.[59] evaluated 60 chronic post-stroke pain patients in a sham-controlled rTMS study. The degree of pain relief was significantly greater for real stimulation than with sham stimulation (22.9% vs 7.8%, respectively). Pain relief was most favorable for fascial pain (trigeminal neuralgia) in the absence of sensory loss, while the effects were not as successful in brainstem stroke.
A meta-analysis study by Leung et al.[60] cited a significant overall analgesic effect with rTMS in comparison to the sham treatment. Moreover, multiple sessions and a lower treatment frequency range appear to generate an improved analgesic outcome.
Vestibular Caloric Stimulation (VCS)
VCS activates the posterior insula, which in turn inhibits the generation of pain in the anterior cingulate gyrus. A single-blind, placebo-controlled study of VCS in 9 patients with CPSP, McGeoch et al. [61] reported a significant immediate treatment effect of cold water caloric stimulation with an average pain reduction of 2.[58] points compared to 0.54 for the placebo group. The authors hypothesized that vestibular function and thermoregulation interaction may be important in maintaining homeostasis, and reset the balance in controlling pain. The patients who responded best to VCS had dominant parieto-insular vestibular cortex (PIVC) located in the non-dominant hemisphere spared from the injury.
Summary
CPSP is a syndrome that occurs after an ischemic or hemorrhagic stroke and can affect tissue anywhere along the spinothalamic pathway, thalamus, or its cortical projections. There are no universally accepted criteria for the diagnosis of CPSP, and the diagnosis is based on the combination of patient history, clinical and neurological findings. The mechanism of CPSP is complex and remains unclear. The management of CPSP is a challenge with no uniformly effective therapies. Therefore, treatment should be individualized. Various pharmacotherapies should be tried initially followed by interventional procedures (e.g., neuromodulation) for the more refractory cases. Improving quality of life and reducing psychological co-morbidity are also essential part of long-term management.
References
- Dejerine J, RoussyG. Le syndrome thalamique. Rev Neurol 1906;14:521-532.
- Andersen G, Vestergaard K, Ingeman-Nielsen M, Jesen TS. Incidence of central post-stroke pain. Pain 1995;61:187-193.
- Widar M, Samuelsson L, Karlsson-Tivenius S, Ahlstrom G. Long-term pain conditions after a stroke. J Rehabil Med 2002;34:165-170
- MacGowan DJ, Janal MN, Clark WC, Wharton RN, Lazar RM, Sacco RL, Mohr JP. Central poststroke pain and Wallenberg’s lateral medullary infarction: frequency, character, and determinants in 63 patients. Neurology Jul 1997;49:120-125.
- Kim JH, Greenspan JD, Coghill RC, Ohara S, Lenz FA. Lesions limited to human thalamic principal somatosensory nucleus (ventral caudal) are associated with loss of cold sensations and central pain. J Neurosci 2007;27:4995-5005.
- Leijon G, Boivie J, Johansson I. Central post-stroke pain – neurological symptoms and pain characteristics. Pain 1989;36:13-25
- Vestergaard K, Nielsen J, Andersen G, Ingeman-Nielsen M, Arendt-Nielsen L, Jensen TS. Sensory abnormality in consecutive, unselected patients with central post-stroke pain. Pain 1995;61:177-186.
- Bowsher D. Central pain: clinical and physiological characteristics. J Neurol Neurosurg Psychiatry 1996;61:62-69.
- Kim JS. Delayed-onset ipsilateral sensory symptoms in patients with central poststroke pain. Eur Neurol 1998;40:201-206.
- Schott GD. Delayed onset and resolution of pain: some observation and implications. Brain 2001;124:1067-1076.
- Goldberger ME, Murray M. Patterns of sprouting and implications for recover of function. Adv Neurol 1988;47:361-85.
- Charriaut-Marlangue C, Aggoun-Zouaoui D, Represa A, Ben-Ari Y. Apoptic features of selective neuronal death in ischemia, epilepsy and gp 120 toxicity. Trends Neurosci 1996;19:109-114.
- Waxman SG, editor. Functional recovery in neurological disease. New York: Raven Press; 1988:157-184.
- Cusick CG. Extensive cortical reorganization following sciatic nerve injury in adult rats versus restricted reorganization after neonatal injury: implications for spatial and temporal limits on somatosensory plasticity. Prog Brain Res 1996;108:379-390.
- Kong KH, Woon VC, Yang SY. Prevalence of chronic pain and its impact on health-related quality of life in stroke survivors. Arch Phys Med Rehabil 2004;85:35-40.
- Misra UK, Kalita J, Kumar B. A study of clinical, magnetic resonance imaging, and somatoensory-evoked potential in central post-stroke pain. J Pain 2008;9:1116-1122.
- Garcia-Larrea L, Convers P, Magnin M, Andre-Obadia N, Peyron R, Laurent B, Mauguiere F. Laser-evoked potential abnormality in central pain patients: the influence of spontaneous and provoked pain. Brain 2002;125:2766-2781.
- Klit H, Finnerup NB, Jensen TS. Central post-stroke pain: clinical characteristics, pathophysiology, and management. Lancet Neurol 2009;8:857-868.
- Ducreux D, Attal N, Parker F, Bouhassira D. Mechanisms of central neuropathic pain: a combined psychophysical and fMRI study in syringomyelia. Brain 2006;129:963-976.
- Craig AD, Chen K, Bandy D, Reiman EM. Thermosensory activation of insular cortex. Nat Neurosci 2000; 3: 184-190.
- Ralston HJ. Pain and the primate thalamus. Prog Brain Res 2005;149:1-10.
- Waxman SG, Hains BC. Fire and phantoms after spinal cord injury: Na+ channels and central pain. Trends Neurosci 2006;29:207-215.
- Wang G, Thompson SM. Maladaptive homeostatic plasticity in a rodent model of central pain syndrome: thalamic hyperexcitability after spinothalamic tract lesions. J Neurosci 2008;28:11959-11969.
- Wasner G, Lee BB, Engel S, McLachlan E. Residual spinothalamic tract pathways predict development of central pain after spinal cord injury. Brain 2008;131:2387-2400.
- Finnerup NB, Johannesen IL, Fuglsang-Frederiksen F, Bach F, Jensen TS. Sensory function in spinal cord injury patient with and without pain. Brain 2003;126:57-70.
- ones AKP, Watabe H, Cunningham VJ, Jones T. Cerebral decreases in opioid receptor binding in patients with central neuropathic pain measured by [11C]diprenorphine binding and PET. Eur J Pain 2004; 8: 479-485.
- Dworkin RH, O’Connor AB, Backonja M, et al. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain 2007;132:237–251.
- Leijon G, Boivie J. Central post-stroke pain—a controlled trial of amitriptyline and carbamazepine. Pain 1989;36:27–36.
- Hansson P. Post-stroke pain case study: Clinical Characteristics, Therapeutic options and long-term follow-up. Eur J Neurol 2004;11 (S1):22-30.
- Lample C, Yazdi K, Roper C. Amitriptyline in the prophylaxis of central poststroke pain. Stroke 2003; 33: 3030-3032.
- Shimodozono M, Kawahira K, Kamishita T, et al. Reduction of central poststroke pain with the selective serotonin reuptake inhibitor fluvoxamine. Int J Neuroscience 2002; 112:1173–1181.
- McQuay H, Carroll D, Jadad AR, Wiffen P, Moore A. Anticonvulsant drugs for management of pain: a systematic review. BMJ 1995;311:1047–1052.
- Vestergaard K, AndersenG, GottrupH, Kristensen BT, JensenTS. Lamotrigine for central poststroke pain: a randomized controlled trial. Neurology 2001;56:184–190.
- Serpell MG. Gabapentin in neuropathic pain syndromes: a randomised, double-blind placebo-controlled trial. Pain 2002;99:557–566.
- Attal N, Brasseur L, Parker F, Chauvin M, Bouhassira D. Effects of gabapentin on the different components of peripheral and central neuropathic pain syndromes: a pilot study. Eur Neurol 1998;40:191–200.
- Vranken JH, Dijkgraaf MG, Kruis MR, van der Vegt MH, Hollmann MW, Heesen M. Pregabalin in patients with central neuropathic pain: a randomized, double-blind, placebo-controlled trial of a flexible-dose regimen. Pain 2008;136:150–157.
- Agnew DC, Goldberg VD. A brief trial of phenytoin therapy for thalamic pain. Bull Los Angeles Neurol Soc 1976;41:9–12.
- Takahashi Y, Hashimoto K, Tsuji S. Successful use of zonisamide for central poststroke pain. J Pain 2004;5:192–194.
- S. Canavero, V. Bonicalzi, and R. Paolotti. Lack of effect of topiramate for central pain. Neurology 2002;58:831-832.
- Attal N, Guirimand F, Brasseur L, Gaude V, Chauvin M, Bouhassira D. Effects of IV morphine in central pain: a randomized placebo-controlled study. Neurology 2002; 58:554–563.
- Rowbotham MC, Twilling L, Davies PS, Reisner L, Taylor K, Mohr D. Oral opioid therapy for chronic peripheral and central neuropathic pain. N Engl J Med 2003;348:1223–1232.
- Bainton T, Fox M, Bowsher D, Wells C. A double-blind trial of naloxone in central post-stroke pain. Pain 1992;48:159–162.
- Iranami H, Yamazaki A, Hatano Y. Tramadol challenge for relief of intractable central poststroke pain. Mayo Clin Proc 2006;81:566.
- Vick PG, Lamer TJ. Treatment of central post-stroke pain with oral ketamine. Pain 2001;92:311–313.
- Yamamoto T, Katayama Y, Hirayama T, et al. Pharmacological classification of central post-stroke pain: comparison with the results of chronic motor cortex stimulation. Pain 1997;72:5–12.
- Sindrup SH, Jensen TS. Efficacy of pharmacological treatments of neuropathic pain: an update and effect related to mechanism of drug action. Pain 1999;83:389–400.
- Attal N, Gaude V, Brasseur L, Dupuy M, Guirimand F, Parker F, et al. Intravenous lidocaine in central pain: a double-blind, placebo-controlled, psychophysical study. Neurology 2000;54:564–574.
- Canavero S, Bonicalzi V, Pagni CA, et al. Propofol analgesia in central pain: preliminary clinical observations. J Neurol 1995;242:561–567.
- Taira T, Kawamura H, Tanikawa T, et al. A new approach to control central deafferentation pain: spinal intrathecal baclofen. Stereotactic Funct Neurosurg 1995;65:101–105.
- Senapati AK, Huntington PJ, Peng YB. Spinal dorsal horn neuron response to mechanical stimuli is decreased by electrical stimulation of the primary motor cortex. Brain Research 2005;1036:173–179.
- Peyron R, Faillenot I, Mertens P, et al. Motor cortex stimulation in neuropathic pain. Correlations between analgesic effect and hemodynamic changes in the brain. A PET study. Neuroimage 2007;34:310–321.
- Nguyen JP, Lefaucheur JP, Decq P, Uchiyama T, Carpentier A, Fontaine D, et al. Chronic motor cortex stimulation in the treatment of central and neuropathic pain. Correlations between clinical, electrophysiological and anatomical data. Pain 1999 ;82:245–251.
- Cruccu G, Aziz TZ, Garcia-Larrea L, et al. EFNS guidelines on neurostimulation therapy for neuropathic pain. Eur J Neurol 2007;14:952–970.
- Fontaine D, Hamani C, Lozano A. Efficacy and safety of motor cortex stimulation for chronic neuropathic pain: critical review of the literature. J Neurosurg 2009;110:251–56.
- Katayama Y, Yamamoto T, Kobayashi K, Kasai M, Oshima H, Fukaya C. Motor cortex stimulation for post-stroke pain:comparison of spinal cord and thalamic stimulation. Stereotact Funct Neurosurg 2001;77:183–186.
- Owen SL, Green AL, Stein JF, Aziz TZ. Deep brain stimulation for the alleviation of post-stroke neuropathic pain. Pain 2006;120:202–206.
- Bittar RG, Kar-Purkayastha I, Owen SL, Bear RE, Green A, Wang S, Aziz TZ. Deep brain stimulation for pain relief: a meta-analysis. J Clin Neurosci 2005;12:515–519.
- Khedr EM, Kotb H, Kamel NF, Ahmed MA, Sadek R, Rothwell JC. Longlasting antalgic effects of daily sessions of repetitive transcranial magnetic stimulation in central and peripheral neuropathic pain. J Neurol Neurosurg Psychiatry 2005;76:833–838.
- Lefaucheur JP, Drouot X, Menard-Lefaucheur I, Zerah F, Bendib B, Cesaro P, Keravel Y, Nguyen JP. Neurogenic pain relief by repetitive transcranial magnetic cortical stimulation depends on the origin and the site of pain. J Neurol Neurosurg Psychiatry 2004;75:612–616.
- Leung A, Donohue M, Xu R, Lee R, Lefaucheur JP, Khedr EM, et al. rTMS for suppressing neuropathic pain: a meta-analysis. J Pain 2009;10:1205-1216.
- Mc Geoch PD, Williams LE, Lee RR, Ramchandran VS. Behavioural evidence for vestibular stimulation as a treatment for central post stroke pain. J Neurol Neurosurg Psychiatry 2008;79:1298–1301.
Leave a commentOrder by
Newest on top Oldest on top